What would you believe?
NMR, MD simulations or BioEMU...
Recently, I encountered some trouble while trying to quantify the population of two states of a very famous and well-studied protein called ubiquitin.
Let’s go back a little…
Ubiquitin is a small protein that post-translationally tags substrates to alter their stability and activity. For the process of “tagging and untagging,” ubiquitin must be highly flexible and dynamic. One of the dynamic motions ubiquitin exhibits is the peptide flip motion. This is the peptide bond rotation of the D52-G53, which can give rise to the two states (IN and OUT). Well, the next obvious question is what are the populations of these two states?

Peptide flip motion showing IN and OUT states
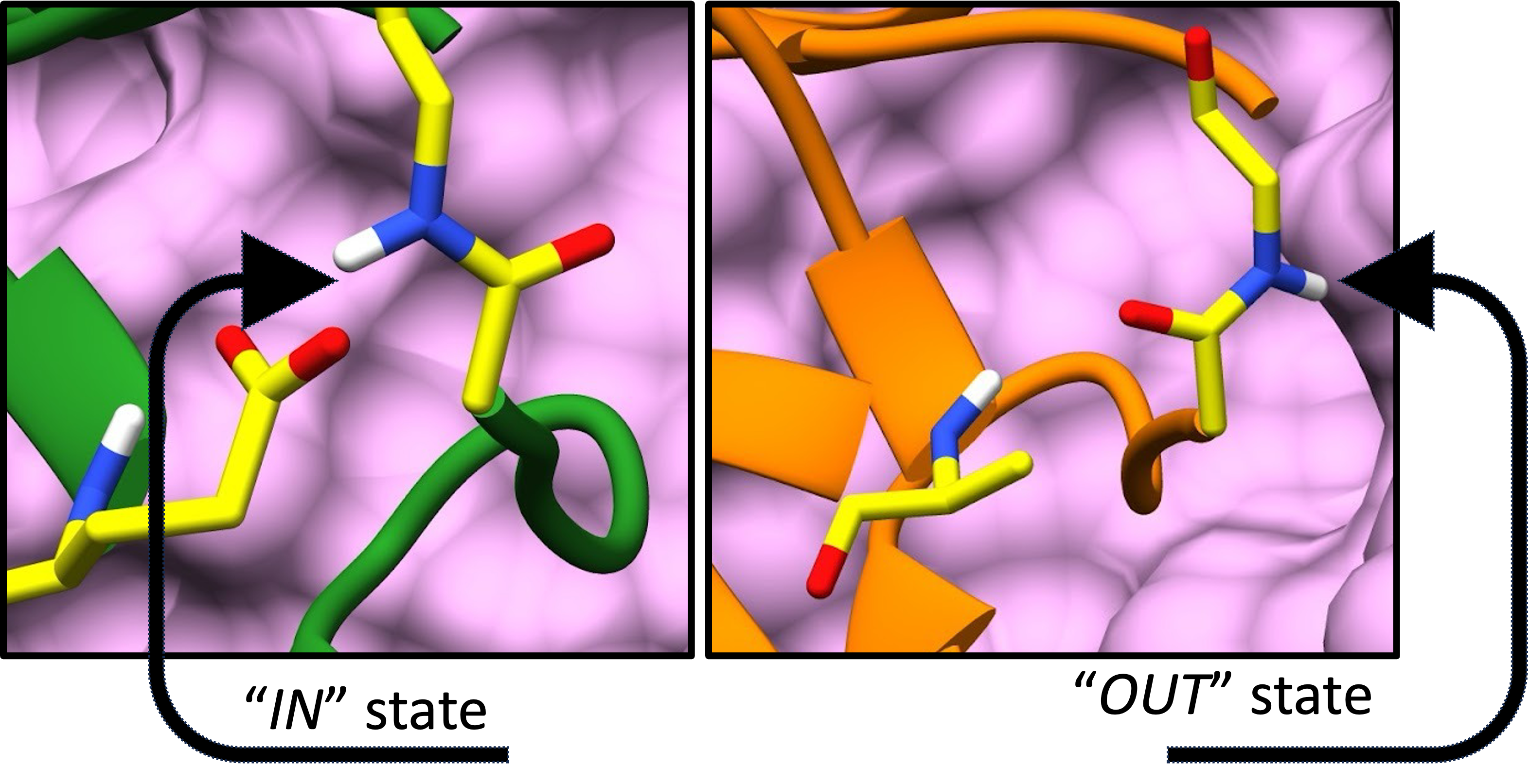
F1: The two conformational states of ubiquitin - IN and OUT
To answer this, I dug a little deep. This dynamic motion occurs in the low-microsecond timescales, making it difficult to probe its kinetics and populations. But there are some results out there that can help!
1. Populations from an NMR study
For this study, the authors locked ubiquitin in either “IN” or “OUT” states by mutations and collected the HSQC-NMR spectrum. The E24 residue shows a shift in its peak position in both the IN and OUT mutants as compared to the wild-type protein. The wild-type peak for E24, shown in red, is right in the center of the IN (black) and OUT (Blue) mutant peaks. This experiment suggests that the peptide-flip motion populates both “IN” and “OUT” states near equally!
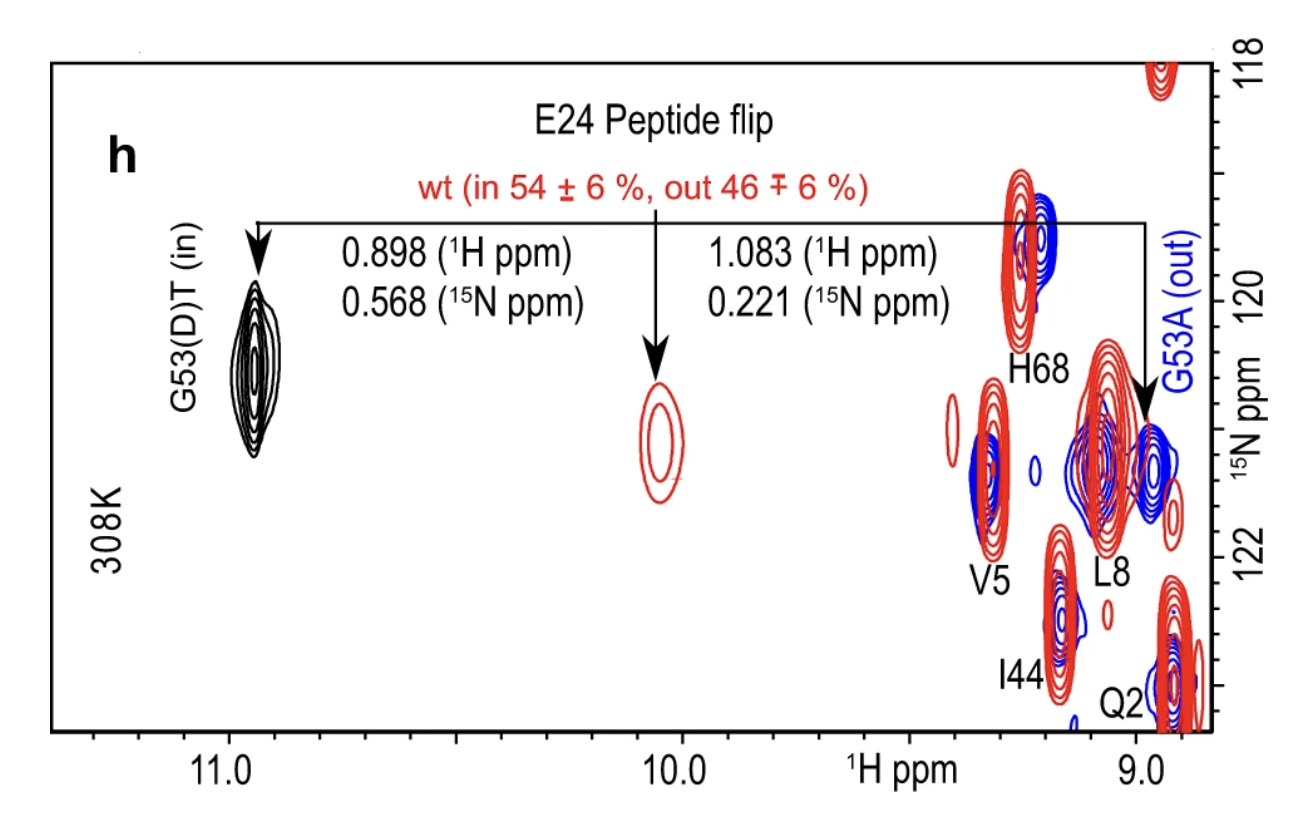
F2: HSQC-NMR spectrum showing E24 peak positions {1}
PIN = 54% and POUT = 46%
2. Populations from MD simulations
Using a two-state Bayesian hidden Markov model, the authors calculated the peptide-flip populations and found that the “OUT” conformation clearly dominates over the “IN” conformation.
PIN = 2% and POUT = 98%
3. Populations from BioEMU
BioEMU is a pretty cool deep learning tool that can be used to sample protein ensembles. You can either run it on Google Colab or even on your local system!
I ran about 10,000 samples for ubiquitin and calculated the populations of “IN” and “OUT” states.
PIN = 3% and POUT = 97%
Now these look like they are in agreement with the MD simulation results.
4. Just for fun: Free and Bound X-ray structures of ubiquitin from the PDB database
Here, I curated about 300 crystal structures and, yet again, calculated the structure in the “IN” vs “OUT” states.
PIN = 20% and POUT = 80%
Now we know conformational selection is a mechanism that various proteins use to bind to selective formations of their partners, and so trying to calculate population from there is not accurate, but I did it for fun anyway!
The NMR results suggest a near equal population of the two states, whereas MD and BioEMU results show a bias towards the “OUT” conformation.
These are some fun discrepancies.
Just some thoughts...
If one were to believe the NMR results, would it mean that BioEMU is not accurate enough for the quantitative study of protein ensembles?
Or are there some artifacts because of which the NMR results are suggesting a near equal population of these two states?
Well, unfortunately, I don’t have these answers yet. If any of you do, please do let me know!
But until then,
Which would you believe?
NMR
MD Simulations
BioEMU
Reference
- A litmus test for classifying recognition mechanisms of transiently binding proteins (2022). {DOI: 10.1038/s41467-022-31374-5}